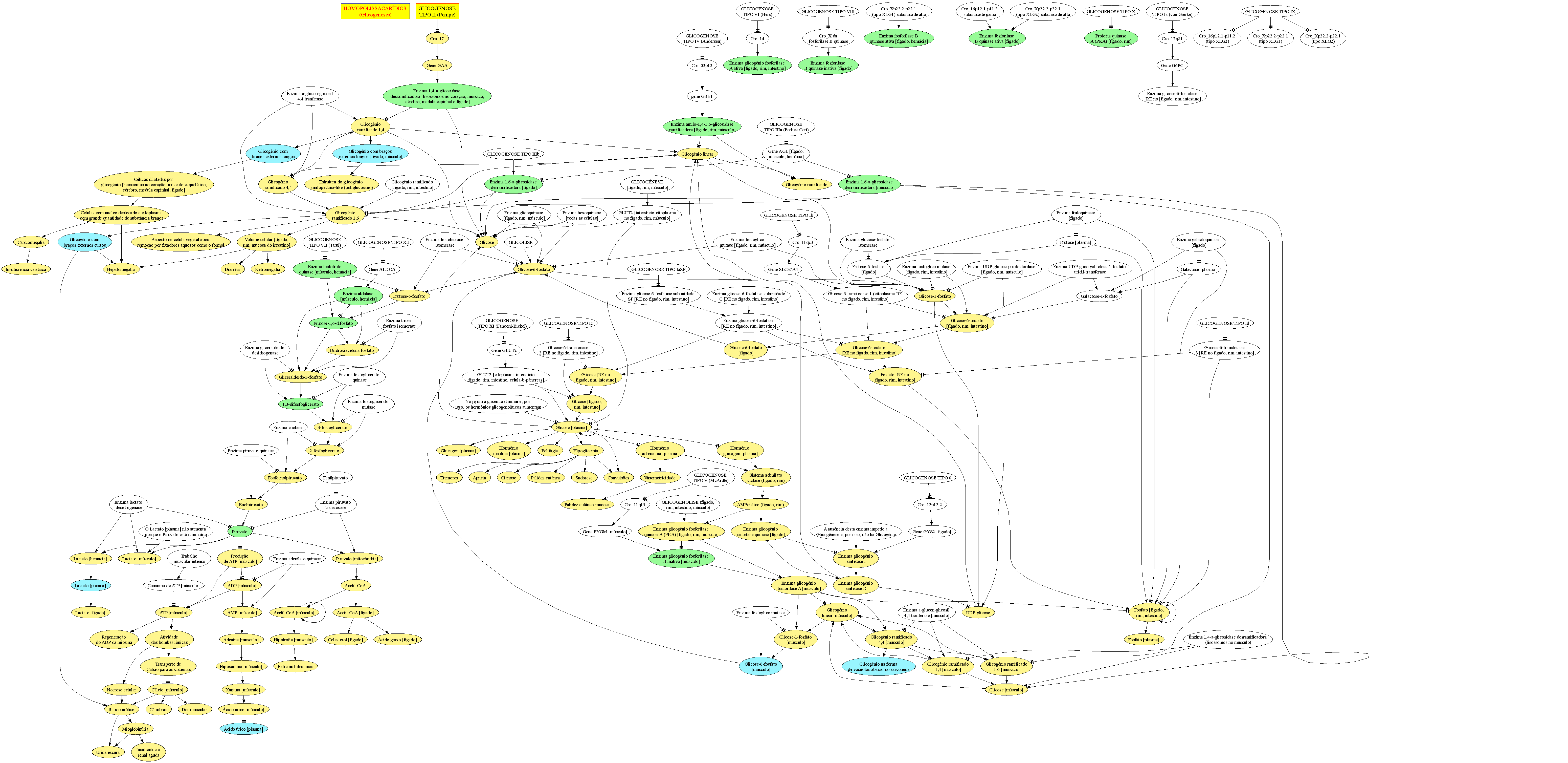
Clínica:1- &.
2- Cardiomegalia, acentuada.
3- Dificuldade de sucção.
4- Dispneia.
5- Esplenomegalia, moderada.
6- Estatura baixa.
7- Evolução crônica, melhora após a puberdade.
8- Face grosseira.
9- Fadiga muscular, às mamadas.
10- Fraqueza.
11- Hepatomegalia, moderada.
12- Hipotonia muscular, difusa.
13- Insuficiência cardíaca.
14- Início gradual, 01M-06M.
15- Macroglossia.
16- Sudorese aumentada, profusa.
17- Taquipneia.